Section 10: UNCERTAINTY QUANTIFICATION FOR ORDINATION ANALYSIS
Pratheepa Jeganathan
04 August, 2021
10_uncertainity_quan_ordination.Rmd
library(phyloseq)
library(tidyverse)
library(genefilter) #KOverA
library(randomcoloR)# distinctColorPalette(n)
library(DirFactor)# Bayesian nonparametric ordination
devtools::load_all()
theme_set(theme_minimal())
theme_update(
text = element_text(size = 10),
legend.text = element_text(size = 10)
)Data
data("psE_BARBI")
threshold <- kOverA(2, A = 25)
psE_BARBI <- phyloseq::filter_taxa(
psE_BARBI,
threshold, TRUE)
psE_BARBI## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 1418 taxa and 86 samples ]
## sample_data() Sample Data: [ 86 samples by 16 sample variables ]
## tax_table() Taxonomy Table: [ 1418 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 1418 tips and 1417 internal nodes ]
ps <- psE_BARBI
rm(psE_BARBI)
ps <- prune_taxa(taxa_sums(ps) > 0, ps)
ps## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 1418 taxa and 86 samples ]
## sample_data() Sample Data: [ 86 samples by 16 sample variables ]
## tax_table() Taxonomy Table: [ 1418 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 1418 tips and 1417 internal nodes ]Edit specimen names
We edit specimen names and identify Asteraceae and non-Asteraceae plants.
sam_names <- str_replace(sample_names(ps), "E106", "E-106")
sam_names <- str_replace(sam_names, "_F_filt.fastq.gz", "")
sam_names <- str_replace(sam_names, "Connor-", "E")
sample_names(ps) <- sam_names
sample_data(ps)$X <- sam_names
sample_data(ps)$unique_names <- sam_names
aster <- c("142","143","15","ST","22","40")
non_aster <- c("33", "71", "106")
paired_aster <- c("E-142-1", "E142-1", "E-142-5", "E142-5", "E-142-10", "E142-10", "E-143-2", "E143-2", "E-143-7", "E143-7", "E-15-1", "E15-1", "ST-CAZ-4-R-O", "ST-CAZ-4-R-M", "ST-SAL-22-R-O", "ST-SAL-22-R-M", "ST-TRI-10-R-O", "ST-TRI-10-R-M")
paired_non_aster <- c("E33-7", "E-33-7", "E33-8", "E-33-8", "E33-9", "E-33-9", "E71-10", "E-71-10","E71-2","E-71-2" ,"E71-3", "E-71-3", "E106-1", "E-106-1", "E106-3", "E-106-3", "E106-4", "E-106-4")
paired_specimens <- c(paired_aster, paired_non_aster)
# We will use 9 colors for 9 different plants
plant_colors <- c("#999999", "#E69F00", "#56B4E9", "#009E73", "#F0E442", "#0072B2", "#D55E00", "#CC79A7", "purple")Bayesian nonparametric ordination
hyper <- list(
nv = 3,
a.er = 1,
b.er = 0.3,
a1 = 3,
a2 = 4,
m = 86, # number of factors - start with one for each specimen (earlier used 22)
alpha = 10,
beta = 0
) # values of the hyper-parameters of priors
# matrix - biological samples are in columns and asvs are in rows
otu <- otu_table(ps) %>%
data.frame() %>%
as.matrix()
#
jabes.mcmc <- DirFactor(
otu,
hyper,
step = 50000,
thinning = 10,# save MCMC results every 10 iterations
save.path = "."
)
jabes.all.res <- lapply(
paste(
"./results",
seq(10010, 50000, 10),
sep = "_"
),
readRDS)
#get bray-curtis distance matrix
all.bc.jabes <- lapply(
jabes.all.res,
function(x){
weights <- x$Q^2*(x$Q>0)*x$sigma
w.norm <- t(weights)/colSums(weights)# normalized Gram matrix
vegdist(
w.norm,
method = "bray"
)# compute BC distance
})
# Use 1000 draws
use.idx <- sample(
1:length(all.bc.jabes),
1000,
replace = F
)
#distatis
sub.bc.ls.jabes <- lapply(
all.bc.jabes[use.idx],
as.matrix
)
plot.bc <- PlotStatis(
sub.bc.ls.jabes,
n.dim = 2,
types = sample_data(ps)$species_names,
dist = T
)
BNO <- plot.bc[[1]] +
scale_color_manual(
values = plant_colors
) +
labs(
col = "Plant type",
x = "Compromise axis 1 (12.4%)",
y = "Compromise axis 1 (7.5%)"
)
ggsave("BNO_all.png", BNO, width = 10, height = 5)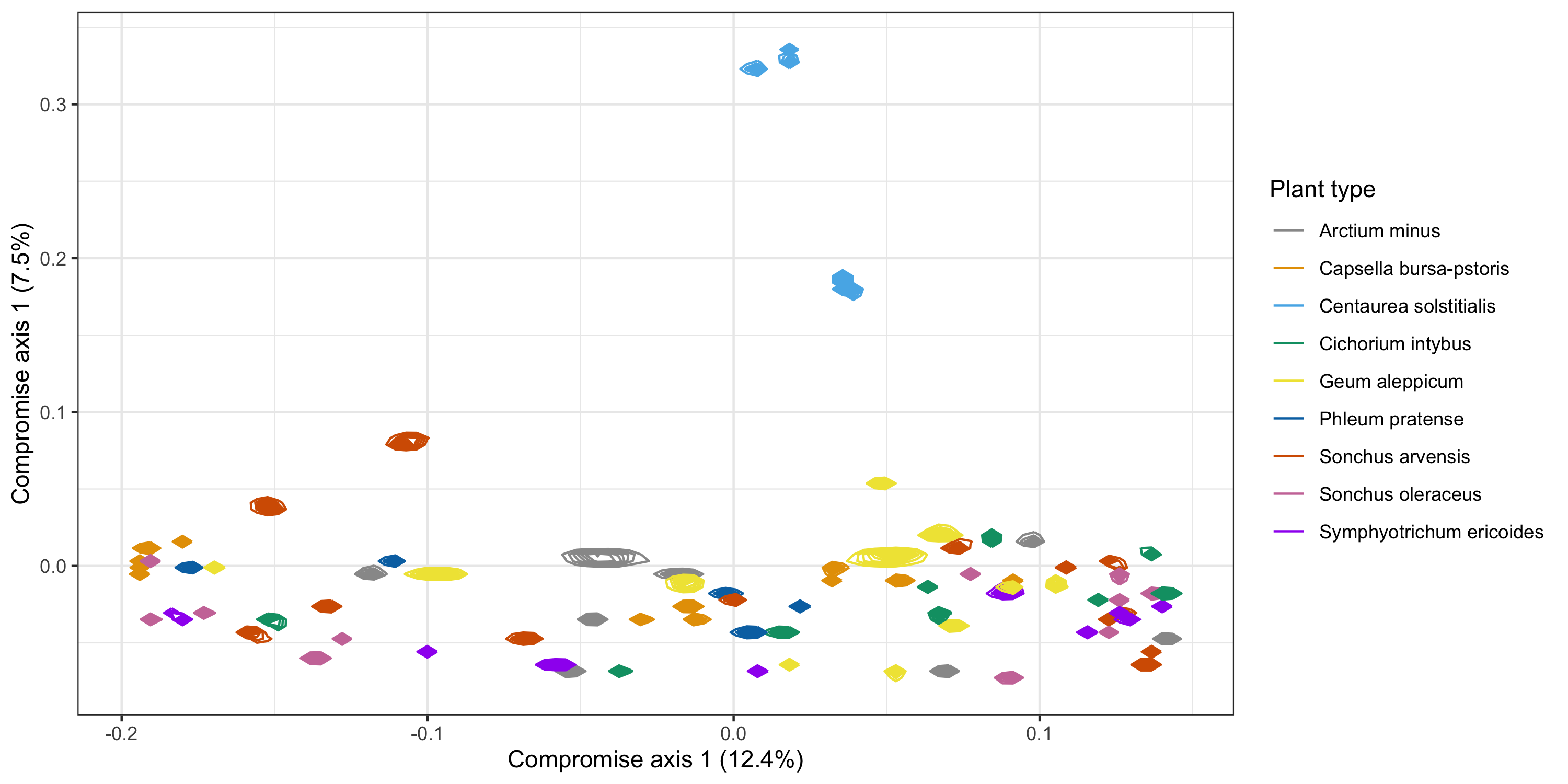
Figure 8: Ordination plot of specimens and 95% posterior credible regions.